Punyaslok Pattnaik received his MS-Dual Degree in Computational Natural Sciences. His research work was supervised by Dr. Deva Priyakumar. Here’s a summary of Punyaslok’s M.S thesis, Machine Learning for Accurate Force Calculations in Molecular Dynamics Simulations as explained by him:
The ability to successfully imitate natural processes is vital for understanding the dynamic behaviour of atoms and molecules. These include reaction rates, mechanisms, energy configurations and macroscopic thermodynamic properties. For any such simulation, it is vital that a potential is constructed which can replicate the dynamics as closely as possible. Widely used classical potentials are constructed by fitting certain parameters with experimental results and then these potentials are used to run simulations under varied conditions. A drawback of this approach is its limited transferability, so the potential might not be suitable to simulate a system under conditions that differ vastly from the parameterization conditions, for example different solvents, temperatures, pressures etc. The accuracy of an empirical force field also depends on the quality of the parameterization that was done based on various approximations and the different sources of data used.
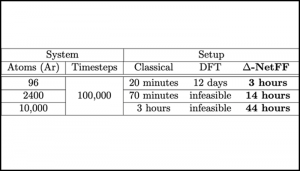
The computationally expensive nature of ab initio molecular dynamics simulations severely limits its ability to simulate large system sizes and long time scales, both of which are necessary to imitate experimental conditions. In this work, we explore an approach to make use of the data obtained using the quantum mechanical density functional theory (DFT) on small systems and use deep learning to subsequently simulate large systems by taking liquid argon as a testcase. A suitable vector representation was chosen to represent the surrounding environment of each Ar atom, and a ∆-NetFF machine learning model where the neural network was trained to predict the difference in resultant forces obtained by DFT and classical force fields was introduced. Molecular dynamics simulations were then performed using forces from the neural network for various system sizes and time scales depending on the properties we calculated. A comparison of properties obtained from the classical force field and the neural network model was presented alongside available experimental data to validate the proposed method.