

Prashanthi Dharanipragada received her doctorate in Bioinformatics. Her research work was supervised by Dr. Nita Parekh and her Ph.D committee panel members included Dr. Vinod Palakkad Krishnanunni (CCNSB, IIIT Hyderabad), Dr. Rahul Siddharthan (IMS, Chennai) and Dr. Sabarinathan Radhakrishnan (NCBS, Bengaluru).
Here’s a summary of Prashanthi’s thesis, Detection and Functional Characterization of Genetic Variations in Diffuse Large B-cell Lymphoma, as explained by her:
Humans share 99.9% of their DNA with each other. This small difference of 0.1% in the DNA sequences is what makes us all unique and comprises thousands to millions of variations termed as ‘Genetic Variations’, ranging from single nucleotide variations to large structural alterations (Figure 1). These genetic variations may directly or indirectly affect the gene expression (gene loss, gene disruption or dosage imbalance) resulting in a modified protein product. Genetic variations majorly cause phenotypic differences amongst individuals. These variations have been shown to play a potential role in predisposition to disease and contribute to variable drug response in individual patients. Hence, it is desirable to study the full spectrum of genetic variations to understand their effects on human health, disease, and evolution.

Figure 1: Types of sequence and structural variations identified in the human genome. A, B, C and D are DNA fragments from the reference and variations of them are represented in the respective category.
Among various types of genetic variations, small sequence variants (SSVs) comprise sequence changes at a single nucleotide level, called single nucleotide variations (SNVs), or a small stretch of DNA sequence (< 50bp) inserted or deleted (INDELs) at a locus. Copy number variations (CNVs) belong to the largest category of structural variations and include duplications, insertions and deletions, varying in size > 50bp to several million bases (Mb). Advancements in next generation sequencing (NGS) technologies have made it possible to carry out variant profiling of individuals, slowly replacing traditional and hybridization-based techniques. However, NGS techniques demonstrate inherent challenges due to short read length, incomplete coverage, GC composition, mappability bias, etc. To address some of these issues, we have developed two open-source, integrated platforms, SeqVItA [1] and iCopyDAV [2], for the detection, functional annotation, and prioritization of SSVs and CNVs, respectively, in NGS data. The reliability of platforms was tested using simulated/real chromosome datasets and are shown to be on par with state-of-art methods. Data pre-treatment step integrated into SeqVItA offers reliable detection of large INDELs (≥10 bp), while the integration of more than one method in various modules of iCopyDAV allows the user to customize the workflow for CNV detection depending on the type and size of CNVs to be detected and the sequencing depth (Figures 2 and 3). The efficacy of the annotation modules, incorporated with various resources, is demonstrated to facilitate personalized medicine based on patient’s mutational landscape.
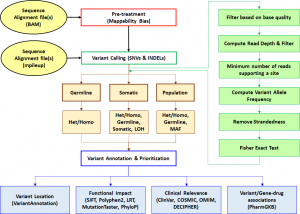
Figure 2: Workflow of SeqVItA for the detection, functional annotation, and prioritization of small sequence variations from NGS data
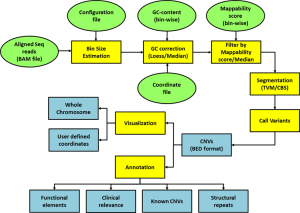
Figure 3: Workflow of iCopyDAV for the detection, functional annotation, prioritization, and visualization of copy number variations from NGS data
Diffuse Large B-cell Lymphoma (DLBCL) is a cancer that originates in B lymphocytes (immune cells) and is one of the most aggressive form of haematological malignancies, with a high incidence rate (~7-8 cases per 100,000). It is characterized by a broad spectrum of genetic aberrations contributing to cancer development and progression. Current treatments based on immunochemotherapy (e.g., R-CHOP) are not completely effective (~40% cases relapse), clearly indicating the presence of subgroups with distinct pathogenesis mechanisms. Based on the cell-of-origin classification, two subgroups of DLBCL, Germinal B cell-like (GCB) and Activated B cell-like (ABC) subtypes are identified at different stages of B cell development and these exhibit different oncogenic pathways and treatment outcomes. Despite several studies carried out to characterize subtype-specific genetic variations, the complete spectrum of these alterations and their relationship with clinicopathological characteristics remains to be elucidated. Detection of subtype-specific genetic variations would assist in understanding the pathogenesis in each case and help in identifying molecular biomarkers for clinical testing, and novel targets for efficient therapy. In this study, we focus on genome-wide analysis of SSVs (SNVs and INDELs) and CNVs in the two subtypes of DLBCL with the objective of identifying molecular markers implicated in the initiation and progression of the tumour in these subtypes. The reason for considering these two categories of genetic variants is that SSVs are widely studied in association studies for disease susceptibility and treatment outcome, while, CNVs affect a large fraction of the genome through focal or arm-length amplifications and deletions contributing to the aggressiveness of cancer. Detection, annotation, and analysis of genetic variations are carried out in 12 DLBCL (7 GCB and 5 ABC subtype) cell lines, using above developed platforms.
Genome-wide detection and analysis of CNVs revealed a varying set of oncogenes and tumour suppressor genes and a few common pathways across cell lines indicating the heterogeneous nature of DLBCL [3]. The pathways involved in immune system responses and NOTCH signalling are affected by CNVs across all the cell lines, while pathways include cell cycle, MAPK, PI3K-AKT-mTOR, JAK-STAT, BCR and NF-κB signalling pathways are affected by CNV-genes in different cell lines. Similar observations are made from the analysis of 48 DLBCL patient samples reported in The Cancer Genome Atlas. This indicated the need for evaluating patient-specific CNV-profile to identify the pathways to be targeted for arresting tumour growth. Based on our analysis, here we propose an in silico genomic-guided approach to identify and target various pathways triggered/disrupted as a consequence of copy number alterations (Figure 4). Detailed analysis of the variant profiles of cell lines also revealed a novel set of recurrent CNVs and associated pathways with a probable subtype-specific role in lymphomagenesis, however, needs to be experimentally confirmed using large data sets with diverse ethnic backgrounds.
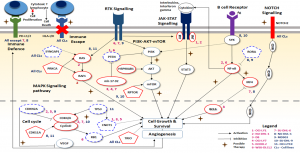
Figure 4: Key immuno-oncogenic pathways affected due to copy alterations in eleven DLBCL cell lines are shown with possible therapeutic targets. Genes in solid and dashed symbols indicate copy gain and copy loss, respectively and novel CNV-genes identified are represented in pentagons.
Analysis of SSVs revealed only a small set (~ 30%) of common variants (≥ 10 cell lines), with the remaining unique to each cell line, indicating molecular heterogeneity in DLBCL. We observed few SSV-genes contributing to variable drug response in individual cell line genomes. Among these, mutations in FCGR3A, FLT4 and CYP2B6 are known to affect the efficacy of drugs Rituximab, Sunitinib, Cyclophosphamide, respectively, which are commonly prescribed in the treatment of DLBCL patients. Functional analysis of oncogenes and tumour suppressors comprising SSVs revealed deregulated cellular processes including impaired immune cells development and differentiation, NOTCH, cell cycle/apoptosis across all the cell lines. Oncogenic pathways such as PI3K-AKT, MAPK and JAK-STAT pathways are observed to be affected in few cell lines. Subtype-specific pathways such as epigenetic modifications (in GCB) and B cell receptor signalling (in ABC) are altered as a result of SSVs in the respective subtype-specific cell lines. The WNT/β-catenin signalling pathway, involved in cancer cell growth, proliferation, and metastasis, is exhibited to be disrupted as a result of SSVs (a feature missed in CNV analysis). We identified many novel SSVs and a few with prognostic significance previously not reported in DLBCL. The precise mechanism through which these mutated genes contribute to DLBCL progression, however, needs to be ascertained.
We next carried out an integrative analysis of SSVs and CNVs identified in DLBCL cell lines. We observe a few immuno-oncogenic pathways affected by various SSVs and CNVs in different key genes. The analysis indicates the heterogeneity in between and within DLBCL subtypes, reflecting varied disease progression and treatment outcome among the patients. Combinatorial analysis of SSV and CNV profiles revealed a comprehensive genomic picture of each DLBCL cell line, demonstrating the need for personalized screening of genetic variations. Finally, integration of SSVs and CNVs uncovered subgroups of shared variations among the cell lines analysed which help in understanding their shared effect on lymphomagenesis and prognosis.
The main contributions of the thesis are:
- Development of an end-to-end platform for detection and functional annotation of sequence variations (SNVs and INDELs) from next generation sequencing data
- Development of an end-to-end platform for detection, functional annotation, and visualization of copy number variations from next generation sequencing data
- Identification and functional characterization of genetic variations in Diffuse Large B-cell Lymphoma cell lines in a subtype-specific manner
- Identification of key immuno-oncogenic pathways disrupted by genetic variations and propose genome-guided in silico therapy that putatively targets these pathways in Diffuse Large B-cell Lymphoma
- Detection of novel recurrent genetic variations with prognostic significance in Diffuse Large B-cell Lymphoma genomes
References:
- Dharanipragada. P., Seelam. S., and Parekh. N., SeqVItA: Sequence Variant Identification and Annotation, Frontiers in Genetics, 2018.
- Dharanipragada. P., Vogeti. S., and Parekh. N., iCopyDAV: Integrated Platform for Copy Number Variations – Detection, Annotation and Visualization, PLOS ONE, 2018.
- Dharanipragada. P. and Parekh. N., Genome-wide characterization of copy number variations in diffuse large B-cell lymphoma with implications in targeted therapy, Precision Clinical Medicine, 2019.