
The new method helps to predict the molecular properties and model chemical reactions.
In groundbreaking research titled ‘Quantum Chemistry Calculations Using Energy Derivatives on Quantum Computers’ that was recently featured in the ‘NISQ Computing Newsletter’ by the USRA Research Institute for Advanced Computer Science, IIITH researcher Utkarsh Azad under the guidance of Prof. Harjinder Singh attempted to showcase the abilities of NISQ hardware for doing quantum chemistry calculations. For the uninitiated, NISQ or Noisy Intermediate-scale Quantum devices refer to the current generation of quantum computers that use the properties of quantum physics to perform calculations, thus outperforming traditional machines such as laptops and others.

Modelling Nature
While explaining his work, Utkarsh begins by quoting Richard Feynman who had famously said, “Nature isn’t classical, dammit, and if you want to make a simulation of nature, you’d better make it quantum mechanical.” The dual degree research student of the Center for Computational Natural Sciences and Bioinformatics (CCNSB) at IIITH goes on to say that modelling chemical reactions on classical computers requires approximations because they can’t exactly calculate the quantum behaviour of more than just a couple of electrons. “The computations are too large and time-consuming,” he remarks, adding that each simplification reduces the overall accuracy of the calculations, thereby limiting their usefulness. Because quantum computing works differently, quantum computers through phenomena known as entanglement, are able to describe electron-electron interactions without approximations. “This means that, at its purest, quantum computing enables us to model nature as it is, without any simplifications,” he says.
What They Did
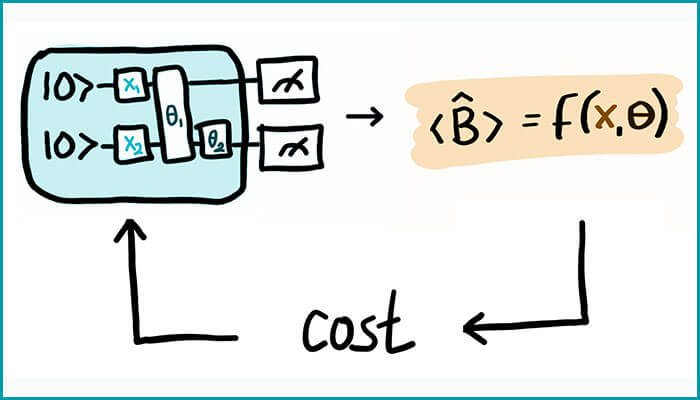
The researchers extended the theory of Variational Quantum Eigensolver (VQE) which is a hybrid quantum-classical algorithm, meaning that it utilizes both quantum and classical computers to perform computations.

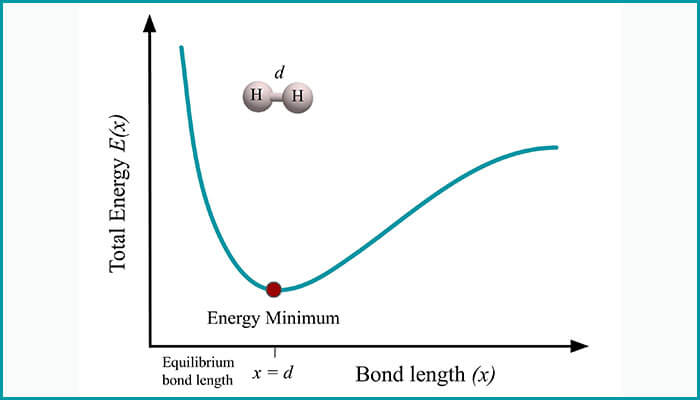
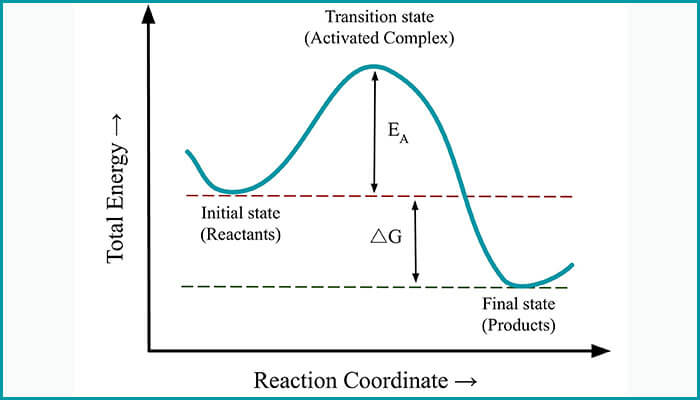
To analyze molecules, not only molecular energies but also the molecular energy derivatives are equally important for calculating a range of time-independent physical and chemical properties. For instance, first-order energy derivatives allow one to search for minimum energy configuration and reaction paths. Utkarsh and Singh’s extension is based on devising a method to calculate molecular energy derivatives for both ground state energy as well as excited state energies up to the second order. They demonstrated how their method could be used on quantum chemistry tasks such as – 1) minimum energy configuration search, 2) estimation of molecular response properties and 3) transition state search.
What Is Unique
Not only are the applications more versatile, but also by doing away with a critical dependency of having to calculate the derivatives of optimal variational parameters, this research scores over previous similar work in energy derivatives. The team also demonstrated how their calculations for molecular response properties significantly improve upon previous results. “To the best of our knowledge, we believe that we are the first group to have shown the usage of NISQ devices in trying to find a stationary point on a potential energy surface, known as transition state structure search (TSS),” says Utkarsh.

Sarita Chebbi is a compulsive early riser. Devourer of all news. Kettlebell enthusiast. Nit-picker of the written word especially when it’s not her own.
It is interesting.. Congratulations to Utkarsh and Dr. Singh. I would like to know more about the minimum energy configuration search using hybrid classical and quantum algorithms
N. V. Suresh Kumar says: